
 Publicar Archivo
Publicar Archivo Reportar Links Dañado
Reportar Links Dañado Cursos Medicos
Cursos Medicos Revistas Medicas
Revistas Medicas Apps Medicas
Apps Medicas
Buscador
 Home
Home Publicar
Publicar Tienda
Tienda Casos Clinicos Nuevo!
Casos Clinicos Nuevo! Imprimir
ImprimirA. Vila Mas, Ll. Puig Sanz
Existen muchas enfermedades que cursan con afectación cutánea y digestiva, algunas de las cuales ya han sido revisadas en otros capítulos (dermatitis herpetiforme, púrpura de Schönlein-Henoch). Nos centraremos en la disqueratosis congénita, síndrome de Peutz-Jeghers, síndrome de Gardner, acrodermatitis enteropática, enfermedad de Wilson y enfermedad de Alagille.
Disqueratosis congénita (síndrome de Zinsser-
Cole-Engman)
Es una enfermedad hereditaria muy poco frecuente que se caracteriza por la presencia de pigmentación cutánea, distrofia ungueal,leucoplasia oral, alteraciones gastrointestinales,oculares, hematológicas y genitales. La mayoría de los casos corresponden a varones y se transmite por una herencia recesiva ligada al cromosoma X (se debe a una mutación del gen que codifica la disquerina, localizado en Xq28); también se ha descrito en mujeres portadoras obligadas, con un patrón de distribución blaschkoide.
Desde el punto de vista clínico, las alteraciones ungueales son habitualmente las primeras en aparecer y consisten en un adelgazamiento de la lámina ungueal con fragmentación fácil,surcos longitudinales con bordes libres irregulares, disminución de tamaño y en algunos casos pueden llegar a desaparecer. Los cambios cutáneos se inician al mismo tiempo o al cabo Es una enfermedad hereditaria muy poco frecuente que se caracteriza por la presencia de pigmentación cutánea, distrofia ungueal, leucoplasia oral, alteraciones gastrointestinales,oculares, hematológicas y genitales. La mayoría de los casos corresponden a varones y se transmite por una herencia recesiva ligada al cromosoma X (se debe a una mutación del gen que codifica la disquerina, localizado en Xq28); también se ha descrito en mujeres
portadoras obligadas, con un patrón de distribución blaschkoide.
Desde el punto de vista clínico, las alteraciones ungueales son habitualmente las primeras en aparecer y consisten en un adelgazamiento de la lámina ungueal con fragmentación fácil,surcos longitudinales con bordes libres irregulares,disminución de tamaño y en algunos casos pueden llegar a desaparecer. Los cambios cutáneos se inician al mismo tiempo o al cabo de pocos años y se caracterizan por una pigmentación fina, reticulada marrón grisácea con atrofia y telangiectasias originando un aspecto poiquilodérmico, más evidente en el cuello, los hombros, la espalda y los muslos (Figs.1 y 2). En la cara hay eritema, atrofia y una pigmentación macular irregular. En el dorso de las manos y los pies la piel es atrófica, fina y brillante y puede asociarse a hiperqueratosis palmoplantar, acrocianosis, hiperhidrosis y ampollas palmoplantares. El pelo de la cabeza,cejas y pestañas es escaso.
Todas las mucosas pueden verse afectadas y la que se altera con mayor frecuencia es la digestiva, especialmente la cavidad oral y la mucosa anal; su presentación puede ser precoz y se caracteriza por pequeñas ampollas, erosiones y áreas de leucoplasia que deben ser controladas periódicamente por el riesgo de degeneración hacia un carcinoma epidermoide. Los dientes suelen ser defectuosos y caen precozmente. Puede producirse estenosis esofágica. En los ojos aparece una obstrucción de los conductos lacrimales con epífora, blefaritis crónica, conjuntivitis y ectropion.
La mayoría de los pacientes presentan alteraciones hematológicas indicativas de fracaso medular (anemia severa, plaquetopenia con diatesis hemorrágica, leucopenia con neutropenia, esplenomegalia y médula ósea hipoplásica) las cuales constituyen la causa de la muerte en un 70% de los casos. Otras manifestaciones son retraso de crecimento posnatal, retraso mental, elevación de las inmunoglobulinas, hemorragia gastrointestinal, ulceración mucosa,calcificación intracraneal, cirrosis hepática,calvicie precoz, cierre incompleto de los arcos
vertebrales, osteoporosis y fragilidad ósea.
También son frecuentes las infecciones oportunistas,sepsis y leucemia. La manifestación genital más característica en los varones es la atrofia testicular.
El diagnóstico es fundamentalmente clínico y suele ser tardío; debe diferenciarse de la anemia de Fanconi (Tabla 1) y de otras enfermedades que cursan con pigmentación poiquilodérmica como el síndrome de Rothmund-Thomson que se transmite de forma autosómica recesiva y se caracteriza por poiquilodermia de inicio más precoz (entre los 3 y 12 meses de vida), predominan en la primera infancia las telangiectasias y la atrofia, fotosensibilidad, cataratas, estatura baja, alopecia, alteraciones dentales e hipogonadismo y no suele haber afectación ungueal ni leucoplasia. También debe diferenciarse de otras entidades que cursen con lesiones blancas de la mucosa oral (leucoedema, liquen plano, enfermedad del injerto contra el huésped, nevo esponjoso blanco, paquioniquia congénita…)
El tratamiento es sintomático y son necesarios controles periódicos para la detección precoz de lesiones malignas. El pronóstico suele ser malo y los principales factores determinantes son el fracaso medular, el desarrollo de neoplasias malignas y las infecciones.
Mas informacion aqui:
Disqueratosis congenita (Zinsser cole Egman)
Síndrome de Peutz-Jeghers
Se caracteriza por la coexistencia de una pigmentación
mucocutánea y poliposis gastrointestinal. Se debe a una mutación en el gen STK 11 (serin-treonin-cinasa 11) del cromosoma 19p13.3 y su transmisión es autosómica dominante aunque se han descrito hasta un 20% de casos esporádicos.
Las lesiones cutáneas pueden estar presentes en el nacimiento o aparecer durante los primeros años de vida y son máculas pigmentadas de color marrón o negro, redondeadas u ovales, de bordes irregulares y menores de 5 mm de diámetro. Las localizaciones más habituales son en los labios, especialmente el inferior, la mucosa oral, las regiones periorbitarias, nasales, palmas y plantas, dorso de dedos y áreas genital, perianal y periumbilical (Figs. 3 y 4). También pueden estar presentes en las encías, paladar duro y lengua. Después de la pubertad las lesiones cutáneas y de los labios tienden a hacerse menos evidentes, aunque las lesiones de la cavidad oral suelen persistir. En la histología existe un aumento de los melanocitos en la capa basal epidérmica con
hiperpigmentación.
Los pólipos gastrointestinales son hamartomas con bajo potencial de malignización. Pueden afectar todo el tracto gastrointestinal, así como las vías respiratorias, biliares y urinarias pero en el 96 % de los casos se localizan en el intestino delgado. Se desarrollan en la segunda década,aunque pueden ser más precoces y aparecer durante la infancia. Las manifestaciones clínicas son ataques recurrentes de dolor abdominal cólico secundarios a la invaginación, anemia
crónica y con menor frecuencia sangrados en forma de rectorragias o hematemesis. La degeneración carcinomatosa es poco frecuente.
El diagnóstico se establece mediante los antecedentes familiares, los hallazgos clínicos y la detección de pólipos hamartomatosos. El diagnóstico diferencial debe realizarse con otras entidades asociadas a lentiginosis (Tabla 2).
El tratamiento de la poliposis debe limitarse a los casos sintomáticos y la técnica de elección es la polipectomía cuando ésta es posible, ya que las resecciones intestinales extensas pueden dejar como secuela una malaabsorción. Se recomiendan controles periódicos mediante contrastes baritados y técnicas endoscópicas.
La supervivencia es similar a la de la población general aunque hay un mayor riesgo de neoplasias, siendo las más frecuentes las de mama,pulmón y gónadas.
Mas informacion aqui:






Síndrome de Gardner
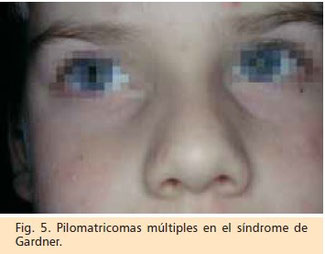
Se considera una variante fenotípica de la poliposis adenomatosa familiar que se asocia a quistes epidermoides con diferenciación pilomatricial, cuya presentación puede ser
precoz (Fig. 5), tumores de tejidos blandos, osteomas y una lesión ocular caracterizada por una hipertrofia congénita del epitelio pigmentario de la
retina (Fig. 6). El gen responsable (APC) se localiza en el cromosoma 5q21-q22 y la enfermedad se transmite de forma autosómica dominante con una
expresividad variable.
Los pólipos se desarrollan entre la 2ª y la 4ª década de la vida aunque pueden aparecer en edades más tempranas y suelen ser asintomáticos. Son de tipo adenomatoso y se localizan en el estómago, intestino delgado y sobre todo en el intestino grueso.Tienen un elevado potencial de transformación maligna que en el 40% de los casos, que se produce al cabo de 15 o 20 años de su desarrollo. Los quistes epidermoides son múltiples y se localizan en la cara, el cuero cabelludo, las extremidades y con menor frecuencia en el tronco; pueden aparecer entre los 4 y 10 años aunque es frecuente un inicio más tardío. Un 50% de los casos presentan osteomas pequeños y múltiples en el maxilar superior e inferior y en el esfenoides; la edad de aparición no está muy clara pero pueden estar ya presentes en la pubertad. También se asocia a exóstosis y alteraciones dentales.
Otras lesiones que pueden estar presentes son pilomatricomas (Fig.6), lipomas subcutáneos, leiomiomas gástricos o retroperitoneales,tumores desmoides y fi brosarcomas. La asociación entre los pilomatricomas y el síndrome de Gardner se postula por la relación metabólica entre la betacatenina, el producto del gen supresor tumoral responsable de los pilomatrixomas y la proteína codificada por el gen APC.
Otras neoplasias asociadas son carcinomas duodenales periampulares y pancreáticos, hepatoblastomas, carcinomas de tiroides y tumores suprarrenales. El diagnóstico es clínico y se realiza por los antecedentes familiares y por la presencia de múltiples quistes epidermoides, que indican la necesidad de realizar una cuidadosa historia familiar, la búsqueda de osteomas y de otros tumores dérmicos. La hipertrofia congénita del epitelio pigmentario de la retina es un hallazgo de gran valor en niños que presentan la mutación y que todavía no han presentado otras manifestaciones. El tratamiento es sintomático.
Mas informacion aqui:



Acrodermatitis enteropática
Es una enfermedad que se trasmite de forma autosómica recesiva (locus génico: 8q24.3) y se debe a una mala absorción de zinc, secundaria al déficit de una proteína transportadora del ión en la mucosa intestinal que está presente en la leche materna. Se caracteriza por la tríada: dermatitis, alopecia y diarrea. El déficit adquirido de zinc, ya sea por aporte insuficiente en la dieta, malaabsorción intestinal o iatrogénico en pacientes con nutrición parenteral, se presenta con un cuadro clínico similar. Los síntomas se inician al cabo de 4 a 6 semanas después del destete o incluso antes si el niño recibe lactancia artificial. Las lesiones cutáneas son vesículoampollosas exudativas, se erosionan y al secarse forman costras localizadas en zonas periorificiales de la cara y periné, rodillas, codos y manos donde pueden adoptar un aspecto psoriasiforme (Fig. 7). Es frecuente la sobreinfección bacteriana y por C. albicans. La alopecia es difusa y pueden afectarse las cejas y las pestañas. La diarrea es el signo más variable pudiendo ser severa y cursar con transtornos hidroelectrolíticos o estar ausente. Puede existir glositis, cambio de coloración del pelo, retraso del crecimiento, fotofobia, irritabilidad y mala cicatrización de las heridas. La biopsia cutánea puede ser útil con fines diagnósticos (Fig. 8).
El diagnóstico es fundamentalmente clínico y puede confirmarse con la determinación de zinc plasmático, que se encuentra por debajo de los valores normales. La fosfatasa alcalina también se encuentra baja por ser un metaloenzima dependiente del zinc. El diagnóstico diferencial es con la dermatitis atópica y con el eccema seborreico infantil. La dermatitis atópica del lactante aparece en los primeros meses de vida, suele haber una historia personal o familiar de atopia (asma, rinitis alérgica, dermatitis) y se manifiesta con eritema, exudación y costras en el cuero cabelludo, las mejillas, el mentón (respetando el triángulo nasolabial), la superficie de extensión de extremidades, las manos y la cara anterior del tronco, se asocia con prurito intenso y evoluciona con brotes recidivantes. El eccema seborreico infantil también aparece en los primeros meses de vida como lesiones eritematoescamosas de bordes policíclicos y defi nidos en el cuero cabelludo (costra láctea), área centrofacial, pliegues cervicales y área del pañal; el niño presenta un buen estado general, no asocia diarrea y gana peso, con un tratamiento tópico adecuado las lesiones mejoran en pocas semanas.
Mas informacion aqui :


El tratamiento con suplementos de zinc oral (2 mg/kg/día) mejora todas las manifestaciones en el plazo de una o dos semanas aunque se requiere un aporte continuo como mínimo
hasta la edad adulta para prevenir las recidivas. En la Tabla 3 se enumeran otras posibles enfermedades carenciales con
manifestaciones dermatológicas.
Enfermedad de Alagille (displasia arteriohepática)
Se caracteriza por la presencia de una hipoplasia biliar intrahepática congénita. Se transmite de forma autosómica dominante (locus 20p12) con una penetrancia casi completa, aunque su expresión clínica es muy variable y puede retrasar el diagnóstico. Son niños que presentan una facies característica (frente prominente, hipertelorismo,
endoftalmos, atrofi a del iris) y a muy corta edad desarrollan colestasis con ictericia y prurito, hipercolesterolemia, xantomatosis, fotosensibilidad e hipogonadismo. Son frecuentes las malformaciones cardiacas (estenosis pulmonar), renales y vertebrales (vértebra en mariposa). En la mayoría de casos la muerte se produce antes de los 5 años, por insuficiencia cardiaca y/o renal.
Mas informacion aqui: enfermedad de Alagille
Envianos tus Artículos o Documentos que deses que aparezcan en nuestra web.| Repetaremos los derechos de Autor, si tu eres el propietario especificalo.
Tienda Piel Sana
Ahora puedes comprar en nuestra pagina web, productos para mantener una piel sana y saludable. y reducir los efectos del envejecimiento.
😎 Envios Nacionales a toda Colombia 
✔ Contamos con metodos de pago PSE, Efecty, Tarjeta de Credito o Mercadopago
Abrir en Nueva Pestaña PielSana
CASOS CLINICOS
Hemos Mejorado la opcion de casos clinicos para que nuestros usuarios puedan interactuar de una manera mas facil, esperamos tu participacion, puedes invitar a amigos al debate..
😎 Para entrar solo coloca un nombre de usuario y listo.
✔ Para agregar un caso tienes que dar click en signo "+" donde dice tesis.
✔ Para comentar un caso solo seleccionalo y dale en el icono de comentario
✔ Si no estas deacuerdo con algo puedes colocar tu comentario en contras o de lo contrario en Pros
Escribir comentario