
 Publicar Archivo
Publicar Archivo Reportar Links Dañado
Reportar Links Dañado Cursos Medicos
Cursos Medicos Revistas Medicas
Revistas Medicas Apps Medicas
Apps Medicas
Buscador
 Home
Home Publicar
Publicar Tienda
Tienda Casos Clinicos Nuevo!
Casos Clinicos Nuevo! Imprimir
Imprimir
Enfermedades mitocondriales y sindromes
INTRODUCCIÓN
En 1962 se describió por primera vez una enfermedad asociada a la transducción de energía mitocondrial. Un paciente de 30 años presentaba un debilitamiento general, transpiración excesiva, alta ingesta calórica sin incremento del peso corporal y un metabolismo basal (una medida de la utilización del oxígeno) excesivamente elevado. Se demostró que el paciente tenía un defecto en el mecanismo que controla la utilización del oxígeno por parte de las mitocondrias. Esta alteración recibió el nombre de enfermedad de Luft. Luft describió hace 40 años el primer error innato del metabolismo energético introduciendo el término “miopatía mitocondrial”, aunque ya se conocían casos que habían sido definidos por sus anomalías bioquímicas consistentes en “acidemia láctica congénita”. La mitocondria tiene un tamaño similar a una bacteria, se reproduce mediante escisión bipartita ycontiene ADN propio que se replica cada vez que el organelo se divide. Su inclusión otorgó a la célula una enorme ventaja selectiva, la capacidad de realizar metabolismo oxidativo, un mecanismo eficiente de generación de energía.
DEFINICIÓN
Las enfermedades mitocondriales son desordenes resultantes de la deficiencia de una o más proteínas localizadas en las mitocondrias e involucradas en el metabolismo. Las enfermedades mitocondriales pueden estar causadas por mutaciones en el ADN mitocondrial, o bien por mutaciones en genes nucleares que codifican para proteínas implicadas en el correcto funcionamiento de la mitocondria.
FISIOPATOLOGIA
Las enfermedades mitocondriales son el resultado del fracaso en el funcionamiento de las mitocondrias, a su vez, partes constitutivas de las células del organismo, cuya función es la de generar la energía necesaria para mantener la vida y para el desarrollo y correcta función de los órganos y sistemas. Cuando fallan, se genera cada vez menos energía en el interior de la célula, produciendo lesión o muerte celular. Si este proceso se repite en todo el cuerpo, los sistemas completos comienzan a fallar y la vida de la persona que lo sufre está en grave riesgo. Esta enfermedad afecta principalmente a los niños, pero los brotes en adultos se están volviendo cada día más frecuente. Las enfermedades de las mitocondrias parecen ocasionar el mayor daño a las células del cerebro, corazón, hígado, músculo, riñón, y a los sistemas endocrinos y respiratorios.
Una enfermedad mitocondrial puede ser un trastorno de múltiples sistemas que afecta más de un tipo de célula, tejido u órgano. Los síntomas exactos no son los mismos para todos, porque una persona con una enfermedad mitocondrial puede tener una mezcla única de mitocondrias sanas y defectuosas, con una distribución única en el cuerpo, además son enfermedades hereditarias, no contagiosas y causadas por mutaciones, o cambios, en los genes. Estos son responsables de construir nuestros cuerpos y son pasados de padres a hijos, junto con cualquier mutación o defecto que tengan. Suelen afectar a miembros de la misma familia de formas diferentes. Para las mitocondrias, hay dos tipos de genes esenciales:
- nADN. Se encuentra dentro del núcleo, la parte de nuestras células que contiene la mayor parte de nuestro material genético o ADN.
- mtADNEI. Reside exclusivamente dentro del ADN contenido en el interior de las mitocondrias.

Las mutaciones, ya sea en el ADN nuclear (nADN) o en el ADN mitocondrial (mtADN), pueden causar enfermedades mitocondriales. A diferencia del nADN, el mtADN pasa únicamente de la madre al hijo. Esto es así porque durante la concepción, cuando la esperma se fusiona con el óvulo, las mitocondrias del esperma y su mtADN son destruidas. Por lo tanto, las enfermedades mitocondriales causadas por mtADN son únicas porque son heredadas en un patrón maternal. Una sola célula puede contener tanto mitocondrias normales como mitocondrias mutantes y el equilibrio entre las dos determinará la salud de la célula. Esto ayuda a explicar por qué los síntomas de las enfermedades mitocondriales pueden variar tanto de persona a persona, aun dentro de la misma familia.
Por qué el mtDNA sufre daño?
El ADNmt es altamente vulnerable a sufrir mutaciones, porque este ADN está muy próximo a los complejos de la cadena y a los radicales libres que éstos generan además de sistemas protectores y de reparación insuficientes.
Se han identificado más de 100 enfermedades asociadas a las mitocondrias, en las que están implicadas una gran variedad de enzimas y sistemas de transporte requeridos para un correcto
mantenimiento y control de la conservación de la energía. Muchas implican el musculo esquelético y el sistema nervioso central. La replicación de la mitocondria depende del DNA mitocondrial
(mtDNA), por lo que más mitocondrias se heredan por vía materna. El ADNmt de un individuo es heredado exclusivamente de su madre, es decir la herencia de las mutaciones del ADNmt no es
mendeliana, una mujer portadora de una mutación mitocondrial puntual la transmitirá a toda su progenie, pero sólo sus hijas la pasarán a sus hijos.Tanto las mutaciones en el mtDNA como
en el DNA nuclear dan lugar a enfermedades genéticas. Las mitocondrias también pueden resultar dañadas debido a la formación de radicales libres (superoxidos) que pueden lesionar el mtDNA.
Así, enfermedades degenerativas relacionadas con la edad, como la enfermedad de Parkinson y de Alzheimer, y las cardiopatías pueden estar relacionadas con lesiones
mitocondriales
MANIFESTACIONES CLINICAS
Algunas manifestaciones multisistemicas de las enfermedades mitocondriales son: retraso desarrollo pondoestatural, fatiga, disnea, retinopatía, hipoacusia, glomerulopatia, síndrome de fanconi, vómitos, diarrea, constipación, endocrinopatías como la diabetes y el hipoparatiroidismo, miocardiopatías, esquizomorfos, anemia sideroblastica, pancitopenia. Además de estas; algunas manifestaciones neurológicas son :trastornos paroxísticos (migraña, epilepsia, sincope, vómitos), trastornos del movimiento (distonías, Parkinson, espasticidad), déficit neurológicos focales (que siguen un perfil vascular), episodios tipo AVE “stroke-like” especialmente en menores de 40 años, encefalopatías recurrentes, demencia, oftalmoplejia (ptosis), síntomas derivados de compromiso de sistema nervioso periférico (miopatía o neuropatía) entre otros muchos. Tabla 1.


TIPOLOGIA DE LOS TRASTORNOS MITOCONDRIALES
Típicamente, estos síndromes son heredados, ya sea en un patrón materno o en un así llamado patrón Mendeliano, y/o son esporádicos, es decir que ocurren sin ningún antecedente hereditario
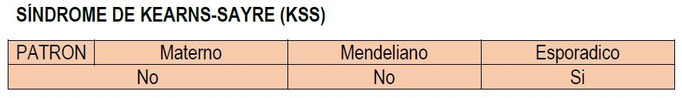
DESCRIPCION
Se caracteriza por su comienzo antes de los 20 años, PEO (Otalmoplejia Externa Progresiva) generalmente como el síntoma inicial y retinopatía pigmentaria, una pigmentación en la retina que puede afectar la visión, pero a menudo la deja intacta. Son síntomas comunes el bloqueo de la conducción cardiaca y ataxia. Menos típicos son el retraso o deterioro mental, maduración sexual atrasada y baja estatura. La enfermedad se presenta de forma esporádica, es decir que ocurre sin ningún antecedente hereditario. El déficit bioquímico suele afectar al complejo IV (citocromo c oxidasa) y prácticamente todos los casos presentan delecciones del mtADN.

DESCRIPCION
Encefalomiopatía necrosante subaguda (MILS = síndrome de Leigh con herencia materna). Se caracteriza por su comienzo durante la infancia. El síndrome de Leigh causa anormalidades cerebrales que pueden resultar en ataxia, convulsiones, visión y oído deteriorados, retrasos en el desarrollo y control alterado de la respiración. También ocasiona debilidad muscular, con dificultad al tragar, en el habla asi como en los movimientos de los ojos.

DESCRIPCION
Síndrome de depleccion del ADN mitocóndrial. Se caracteriza por su comienzo durante la infancia. Este trastorno típicamente causa debilidad muscular y/o fallo hepático y, más raramente, anormalidades cerebrales. La debilidad, las dificultades de alimentación y los retrasos en el desarrollo son síntomas comunes; la PEO y las convulsiones son menos comunes.

DESCRIPCION
Encefalomiopatía mitocondrial con acidosis láctica y episodios que simulan ictus. La enfermedad tiene una transmisión materna en la mayoría de los casos publicados. La edad de comienzo de la enfermedad es variable aunque la mayoría de los enfermos debutan antes de los 15 años, presentando cefaleas de carácter migrañoso, a las que se acompaña un cuadro deficitario cerebral que simula un infarto, generalmente en forma de ceguera cortical, hemianopsia o afasia. Al primer episodio de ictus se suceden otros, siendo la recuperación incompleta y llevando finalmente al enfermo a un importante deterioro neurológico. Pueden existir crisis comiciales. Los síntomas musculares suelen consistir en debilidad muscular e intolerancia al ejercicio, u oftalmoplejia. Otros síntomas comunes incluyen PEO, debilidad muscular generalizada, intolerancia al ejercicio, pérdida del oído, diabetes y baja estatura. En casi todos los enfermos se aprecian RRF en la biopsia muscular. La acidosis láctica es frecuente y en los estudios de TAC destaca la existencia de lesiones de aspecto isquémico (hipodensidades córtico-subcorticales), en distintos estadios evolutivos y que no corresponden a áreas vasculares precisas. No existe tratamiento eficaz para este trastorno. Más de la mitad de los pacientes presentan déficit de la cadena respiratoria mitocondrial, que casi siempre afecta al complejo I . La mayoría de los pacientes presentan mutaciones puntuales del mtADN que afectan al gen que codifica el ARN de transferencia para la leucina (tARNLeu).

DESCRIPCION
Epilepsia mioclónica con fibras rojas rasgadas. Se caracteriza por comenzar tarde en la niñez a adolescencia. Los síntomas más prominentes son mioclonias (contracciones musculares), convulsiones, ataxia y debilidad muscular. La enfermedad también puede causar pérdida del oído y baja estatura. Se define por la asociación de epilepsia mioclónica y la aparición en la biopsia muscular de fibras RRF. Las primeras manifestaciones clínicas (casi siempre las mioclonías) suelen aparecer en la adolescencia y suelen asociarse a otros tipos de crisis comiciales, tales como atónicas o tónico-clónicas. El cuadro clínico global es variado, de forma que a las mioclonías se pueden unir, en distinta proporción, un síndrome cerebeloso, debilidad muscular con fatigabilidad excesiva, hipoventilación central, crisis generalizadas, demencia y otras. El que lleva a la muerte en pocos años, a otros con incapacidad funcional leve. La biopsia muscular muestra (por definición) RRF, pero son más frecuentes las fibras con déficits focales de COX. El láctico sérico suele estar elevado, sobre todo durante el ejercicio. El EEG puede mostrar un enlentecimiento difuso sobre el que emergen anomalías comiciales. En la TAC y RMN craneal se encuentran atrofia de cerebelo, tronco y hemisferios cerebrales. Los hallazgos bioquímicos descritos son variables, pero lo más frecuente es el déficit de complejo IV. La enfermedad se hereda de forma materna, siendo característica la gran variedad de síntomas presentes en la misma familia; los estudios genéticos demuestran en la mayoría de los casos una mutación puntual del gen que codifica el ARN transferente de la lisina (tARNLys).

DESCRIPCION
Encefalomiopatía neurogastrointestinal mitocóndrica. Inicio: usualmente antes de los 20 años de edad. Características: Este trastorno causa PEO, ptosis, debilidad en las extremidades y problemas gastrointestinales (digestivos) incluyendo diarrea crónica y dolores abdominales. Otro síntoma común es la neuropatía periférica (una disfunción de los nervios que puede llevar a reducción sensorial y debilidad muscular). Típicamente la RNM cerebral de estos enfermos muestra una importante afectación de la sustancia blanca (leucoencefalopatia)

DESCRIPCION
Neuropatía, ataxia y retinitis pigmentosa. Inicio: infancia a edad adulta. Características: NARP causa neuropatía (ver arriba), ataxia y retinitis pigmentosa (degeneración de la retina en el ojo, resultando en pérdida de la visión). También puede ocasionar retraso en el desarrollo, convulsiones y demencia

DESCRIPCION
Suele iniciarse en la infancia. Aparece de forma esporádica, es decir, este síndrome causa anemia severa y disfunción del páncreas. Los niños que sobreviven esta enfermedad usualmente pasan a desarrollar KSS.

DESCRIPCION
Oftalmoplegia externa progresiva. Usualmente, se inicia en la adolescencia o edad adulta temprana. La PEO es a menudo un síntoma de enfermedad mitocondrial, pero algunas veces se destaca como un síndrome determinado. A menudo está relacionado con intolerancia al ejercicio.
MIOPATÍA
Los principales síntomas de la miopatía mitocondrial son debilidad muscular e intolerancia al ejercicio. Es importante recordar que la severidad de cualquiera de estos síntomas varía enormemente de una persona a otra, aun dentro de la misma familia. La debilidad muscular es habitualmente más frecuente en los músculos que controlan los movimientos de los ojos y los párpados y entonces se diagnostica de Miopatía Ocular.
También pueden ocasionar debilidad en otros músculos de la cara y el cuello, lo que puede llevar a problemas de pronunciación y dificultad al tragar. En estos casos, pueden ser útiles la terapia del habla, logopedia o los cambios en la dieta con comidas más fáciles de tragar. Algunas veces las personas con miopatías mitocondriales experimentan pérdida de la fuerza muscular en los brazos o las piernas y pueden necesitar aparatos ortopédicos o sillas de ruedas para movilizarse. La intolerancia al ejercicio, se refiere a sensaciones extraordinarias de agotamiento como resultado de un esfuerzo físico. El grado de intolerancia al ejercicio varía enormemente entre individuos. Algunas personas pueden tener problemas solamente con actividades atléticas como correr, mientras que otras pueden experimentar problemas con actividades cotidianas como caminar o levantar un peso de un kilo.
A veces, la intolerancia al ejercicio se puede acompañar de calambres musculares dolorosos en los músculos que han realizado un esfuerzo fisico. Ocurren cuando alguien con intolerancia al
ejercicio "se excede" y pueden aparecer durante el ejercicio, o varias horas más tarde.








ENCEFALOMIOPATÍA
Una encefalomiopatía mitocondrial típicamente incluye algunos de los síntomas de miopatía mencionados al lado, más uno o más síntomas neurológicos y de otros organos. De nuevo, estos síntomas demuestran una gran variabilidad individual, tanto con relación a su tipo como a su severidad. Algunos de los síntomas comunes de una encefalopatía mitocondrial son las jaquecas tipo migrañas, convulsiones, pérdida auditiva y con frecuencia ataxia. En por lo menos un síndrome, las jaquecas y convulsiones se presentan acompañadas de apoplejía (interrupción del suministro de sangre a una zona del cerebro).
Afortunadamente, existen buenos tratamientos para algunas de estas condiciones. Las pérdidas auditivas pueden manejarse utilizando aparatos para sordos y formas alternativas de comunicación. A menudo, las jaquecas pueden aliviarse con medicamentos y las convulsiones pueden prevenirse con drogas utilizadas para la epilepsia (fármacos antiepilépticos). Actualmente existen varias drogas que se están investigando para el tratamiento de apoplejías. Para la ataxia, o problema de equilibrio y coordinación, se pueden tratar con terapia física y ocupacional así como aparatos para mantener la postura. En los casos más graves se acude al uso de silla de ruedas.
Además de afectar la musculatura del ojo, una encefalomiopatía mitocondrial, puede afectar el ojo en sí y partes del cerebro involucradas en la visión (Por ejemplo, las cataratas o lesiones cerebrales focales).
·
Otros síndromes multisistemicos asociados con alteración mitocondrial son:
- Epilepsia Mioclónica con FibrasRojas Rasgadas (MERRF)
- MELAS (Encefalomiopatía Mitocondrial con Acidosis Láctica y episodiostipo Accidentes Vasculares Cerebrales)
- Oftalmoplejia externa progresiva crónica (CPEO)
- Síndrome de Kearns-Sayre (SKS)
- Neuropatía Óptica Hereditariade Leber (LHON)
- Neuropatía, Ataxia y RetinitisPigmentosa (NARP)
- Síndrome de Wolfram
- Síndrome Mioneurogastrointestinal (MNGIE)
- Sindrome de Leigh o EncefalopatíaNecrotizante Subaguda
- Deficiencia del Complejo I
- Deficiencia del complejo II
- Deficiencia del complejo III
- Deficiencia del complejo IV
- Deficiencia del complejo V
- Trastornos por depleción del ADNmt
DIAGNOSTICO

La extremada variabilidad en los síntomas de las enfermedades mitocondriales hace que el diagnóstico sea, en ocasiones, difícil. Desde el punto de vista clínico, se ha de sospechar la existencia de una enfermedad mitocondrial, ante la presencia de los signos clínicos de entidades concretas y ya descritas; también se ha de sospechar cuando aparezca una “asociación inexplicable de síntomas” en el sentido de la coexistencia de lesión de diferentes tejidos ú órganos no relacionados funcional o embriológicamente.
Existen diferentes criterios utilizados para el diagnóstico de las enfermedades mitocondriales con diferentes procedimientos:
-
Examen clínico:
Exploración neurológica.
Balance muscular.
Test de esfuerzo para determinar la producción de lactato.
- Biopsia muscular que puede mostrar anomalías bastante específicas de patología mitocondrial como son las fibras rojo-rasgadas (RRF), aumento de la actividad periférica con SDH y fibras COX negativas. También pueden encontrase mitocondrias anormales en microscopia electrónica.
- Estudio bioquímico de la biopsia muscular para determinar déficits de los complejos de la cadena respiratoria.
- Estudio genético molecular para identificar anomalías en el ADN mitocondrial.
TRATAMIENTO
Todos los tratamientos ensayados aportan actualmente escasos beneficios para mejorar la disfunción mitocondrial. Las enfermedades mitocondriales pueden expresarse de formas muy diversas, afectando a cualquier tejido, a cualquier órgano y en cualquier momento de la vida. No existen series largas de pacientes con el mismo defecto molecular y la misma manifestación clínica que permitan realizar estudios concluyentes sobre la efectividad de los diversos fármacos aplicados en el tratamiento. Los tratamientos sólo se han mostrado útiles en un número limitado de pacientes, mientras que en la gran mayoría de ellos las medidas terapéuticas se limitan a ser de soporte.
El tratamiento de las enfermedades mitocondriales ha de ser necesariamente multidisciplinario para evaluación y tratamiento de las alteraciones cardiacas, sensoriales, endocrinas, ortopédicas, etc. Agruparemos el tratamiento de estas enfermedades en los siguientes apartados:
- Terapia farmacológica específica: Se pueden paliar los efectos de estas enfermedades a través de tratamientos farmacológicos. Los siguientes fármacos son de uso común en patologías mitocondriales Carnitina, Coenzima Q, Dicloroacetato, Monohidrato de carnitina, Prednisona, Riboflavina, Vitamina C, Vitamina E, Vitamina K3.
- Terapia genética preimplantacional: Trasplante nuclear de óvulos in vitro, fertilización in vitro de donación de óvulos. Se realizan técnicas de preimplantación genética, en madres portadoras de mutaciones del ADNmt. Consisten en un transplante del núcleo a un ovocito de una donante y a continuación hacer una fecundación in vitro con esperma paterno.
- Tratamiento de mantenimiento: Se deben disponer una serie de cuidados generales en el control evolutivo de estos pacientes según el órgano afectado. Se considera apropiado seguir un tratamiento de rehabilitación y de fisioterapia. En algunos casos también se recomienda recibir terapia de logopeda.
- Tratamiento psicológico: En determinados casos y familias se recomienda apoyo psicológico y en ocasiones psiquiátrico.
FÁRMACOS CON RIESGO DE TOXICIDAD PARA LA MITOCONDRIA
- Antibióticos: tetraciclina, ciprofloxacina, aminoglicosidos (en pacientes con la mutación A1555G).
- Antivirales: Azydothymidina (AZT). Fialuridine y drogas que deplecionan al ADNmt en pacientes con depleción del mismo.
- Antiepilépticos: especialmente el valporato sódico, que inhibe la fosforilación oxidativa y afecta a la oxidación de los ácidos grasos. También se deben evitar los barbitúricos y las hidantoinas.
- Anestésicos: se debe evitar la administración de etomidato y thiopentona en el síndrome de Kearns–Sayre. En los pacientes con enfermedad mitocondrial se debe tener en cuenta la gran sensibilidad al antracurium y roncuronium. Algunos autores han descrito complicaciones con la utilización de Fentanilo y Thiopental.
Los pacientes con afectación mitocondrial presentan mayor riesgo de fallo respiratorio en el
postoperatorio quirúrgico debido en parte a la mayor producción de citoquinas y la consiguiente formación de óxido nítrico que en grandes cantidades afecta la producción de energía. Se aconseja
siempre que sea posible la utilización de anestesia epidural. También se debería evitar la hipoxia crónica, que pueden provocar anestesias irreversibles.
Envianos tus Artículos o Documentos que deses que aparezcan en nuestra web.| Repetaremos los derechos de Autor, si tu eres el propietario especificalo.
Tienda Piel Sana
Ahora puedes comprar en nuestra pagina web, productos para mantener una piel sana y saludable. y reducir los efectos del envejecimiento.
😎 Envios Nacionales a toda Colombia 
✔ Contamos con metodos de pago PSE, Efecty, Tarjeta de Credito o Mercadopago
Abrir en Nueva Pestaña PielSana
CASOS CLINICOS
Hemos Mejorado la opcion de casos clinicos para que nuestros usuarios puedan interactuar de una manera mas facil, esperamos tu participacion, puedes invitar a amigos al debate..
😎 Para entrar solo coloca un nombre de usuario y listo.
✔ Para agregar un caso tienes que dar click en signo "+" donde dice tesis.
✔ Para comentar un caso solo seleccionalo y dale en el icono de comentario
✔ Si no estas deacuerdo con algo puedes colocar tu comentario en contras o de lo contrario en Pros
Escribir comentario